Neu entdeckter Gendefekt stört Blutbildung und Immunsystem
(Wien, 21.06.2023) Auf der Suche nach dem Ursprung der rätselhaften Symptome von vier Kindern haben Forscher:innen der St. Anna Kinderkrebsforschung, des CeMM Forschungszentrum für Molekulare Medizin der ÖAW und der MedUni Wien gemeinsam mit Kooperationspartner:innen Störungen der Blutbildung, des Immunsystems und Entzündungen in Zusammenhang gebracht und dabei eine völlig neue Erkrankung entdeckt. Diese bahnbrechende Entdeckung liefert die Grundlage dafür, dass ähnliche Krankheiten besser verstanden werden. Ein Meilenstein, den die Forscher:innen jetzt in einer weltweit hoch angesehene Fachzeitschrift, dem New England Journal of Medicine, veröffentlichen konnten.
Ein neu entdeckter Defekt im DOCK11 Gen führt sowohl zu Störungen der weißen als auch der roten Blutkörperchen. „Solche seltenen und bisher unbekannten Erkrankungen eröffnen uns wertvolle Einblicke in die grundlegenden Prinzipien der Blutbildung und des Immunsystems. Sie ermöglichen uns ein besseres Verständnis der fehlregulierten Prozesse, die diesen Erkrankungen zugrunde liegen“, erklärt Univ.-Prof. Dr. Kaan Boztug, Letztautor der Studie und Wissenschaftlicher Direktor der St. Anna Kinderkrebsforschung.
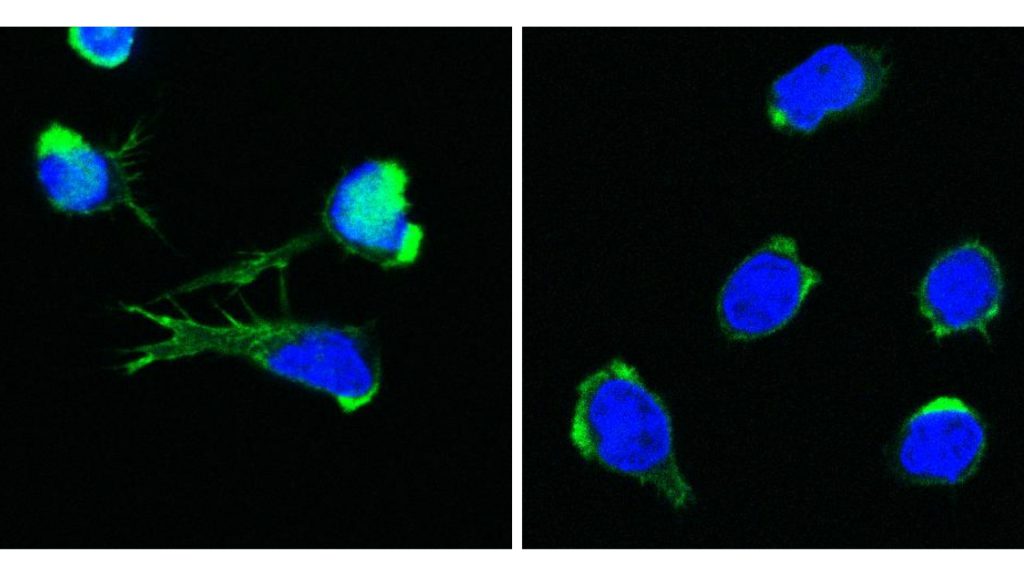
Erstmals erkannt wurde der Gendefekt bei einem jungen Patienten aus Spanien, bei dem trotz wiederholter Tests keine Erklärung für die schwere Entzündung von diversen Organen wie Niere, Darm und Haut gefunden werden konnte. Der behandelnde Arzt wandte sich daher mit Blutproben des Patienten an die St. Anna Kinderkrebsforschung. Und hatte Erfolg. Das international führende Zentrum für seltene Erkrankungen der Blutbildung und des Immunsystems fand die Ursache. „Wir arbeiten eng mit internationalen Partnerinstitutionen zusammen, um solche Krankheiten zu verstehen und den Patient:innen zu helfen“, sagt Boztug, der auch Professor im Fachbereich Kinderheilkunde und Entzündungsforschung an der Medizinischen Universität Wien und Adjunct Principal Investigator am CeMM Forschungszentrum für Molekulare Medizin der ÖAW ist.
Drei weitere Patienten mit Varianten im gleichen Gen
Die Genomsequenzierung des Patienten enthüllte einen schwerwiegenden Defekt im Gen DOCK11, einem Gen, das bisher nicht mit einer menschlichen Erkrankung in Verbindung gebracht wurde und an der Kommunikation zwischen Zellen beteiligt ist. „Wir hatten zu Beginn nur einen Patienten und wussten daher nicht, welche Symptome im Zusammenhang mit diesem Gendefekt stehen und welche nur zusätzliche Begleiterscheinungen sind. Das war eine ziemliche Herausforderung“, sagt Jana Block, MSc, Erstautorin der Publikation und PhD Studentin in Boztugs Forschungsgruppe an der St. Anna Kinderkrebsforschung und am CeMM. Den Wissenschafter:innen gelang es in der Folge durch ihre internationale Vernetzung, weitere Patienten mit ähnlichen DOCK11 Mutationen zu finden und damit ein klareres Bild der Krankheit zu bekommen.
DOCK11 kontrolliert Blutbildung
Einer der Patienten litt unter einer stark verminderten Anzahl roter Blutkörperchen und benötigte regelmäßige Bluttransfusionen. „Bei Immundefekten kommt es häufig zu Anämie, und oft werden die eigenen roten Blutkörperchen durch Autoantikörper zerstört, die sich gegen die eigenen Blutzellen richten. Im gesunden Menschen passiert das aber nicht“, erklärt Jana Block. „In unseren Untersuchungen haben wir jedoch keine solchen Autoantikörper gefunden.“ Die Ursache für die verminderte Anzahl von roten Blutkörperchen klärten die Wissenschafter:innen dann mithilfe eines Zebrafischmodells auf – der DOCK11 Defekt führte zu einer gestörten Bildung der Blutkörperchen und damit zu einem neuartigen Mechanismus für Anämie, einem Mangel an roten Blutkörperchen.
DOCK11 hält T-Zellen in Schach
Im Menschen war die Rolle von DOCK11 bisher unbekannt, vorherige Studien hatten eine Bedeutung des Proteins für die Entwicklung der B-Zellen in Mausmodellen gezeigt. Die Forscher:innen konnten nunmehr zeigen, dass sich auch in Patienten mit DOCK11 Defizienz manche B-Zellen nicht richtig entwickeln und gleichzeitig auch andere Immunzellen – die sogenannten T-Lymphozyten – überaktiviert sind. „Das Protein scheint eine Rolle dabei zu spielen, die Aktivierung der T-Zellen in einem bestimmten Rahmen zu halten“, erklärt Block. Das sei wichtig, da eine Überaktivierung zu Schäden an umliegenden Geweben und Organen führen könne, besonders wenn gar kein Erreger vorhanden sei. Ein Zusammenhang zwischen T-Zell-Defekten und Prädisposition für Tumorerkrankungen wird bei anderen Immundefekten beobachtet und kann daher auch bei DOCK11 Defizienz nicht ausgeschlossen werden. „Dieser Frage werden wir jetzt in unseren Forschungsarbeiten natürlich noch weiter nachgehen“, sagt Boztug.
Stammzellentransplantation als mögliche Therapieform
Obwohl noch immer nicht alle Details der Funktion von DOCK11 verstanden sind, vermuten die Forscher:innen, dass die Transplantation von blutbildenden Stammzellen die Erkrankung heilen könnte. „Natürlich müssen diese Fragen in zukünftigen Studien entsprechend untersucht werden. Es ist auch denkbar, dass man DOCK11 Defizienz mittels Gentherapie behandeln könnte“, meint Boztug. Dass es nun Hoffnung auf die Entwicklung von Therapien gibt, hat für die Wissenschafter:innen eine ganz besondere Bedeutung: „Unsere Arbeit der letzten fünf Jahre ist zweifellos ein bedeutender Meilenstein, jedoch haben wir auch im Blick, dass mehrere der Patienten an der Erkrankung verstorben sind“, sagt Block. Die Forscherin hofft, dass nun weitere Zentren auf diese Genmutation aufmerksam werden und die Erforschung der Landkarte dieser Krankheit damit weiter vorantreiben. „Diese Erkrankung ist wirklich schwerwiegend, deshalb sehen wir es als unseren Auftrag, nicht nur die biologischen Prozesse zu verstehen, sondern auch darauf basierend wirksame Therapiestrategien zu entwickeln“, ergänzt Boztug.
 für Wordpress und WooCommerce.")
